




In a single day, enough sunlight strikes Earth to power the world for an entire year—that is, if we can find a way to capture that energy cheaply and efficiently. While the cost of solar energy has decreased dramatically, current silicon-based solar cells are expensive and energy-intensive to manufacture, prompting researchers to look for alternatives.
Perovskite solar cells are a prime contender for the next generation of this renewable energy. These synthetic materials are cheaper and require less energy to produce but fall behind many silicon-based cells in terms of their stability and efficiency. Now, a paper published in Nature Communications from the groups of the University of Pennsylvania’s Andrew M. Rappe and Yueh-Lin Loo of Princeton University gives insight into how the molecular make up of certain perovskites might affect their efficiency and offers a path forward to better solar cells using a simple metric.
“The world currently needs more efficient and cost-effective photovoltaic cells, and 3D hybrid perovskite PVs have taken the world by storm,” says Rappe, a professor in Penn’s Department of Chemistry who also co-directs Penn’s VIPER program. “But they are irreversibly damaged by water, which is a showstopper for practical applications. Inserting organic molecular planes between 2D hybrid perovskite planes is a promising scheme to provide efficient, low-cost, and robust solar cells.”
In this study, the researchers investigated a certain class of perovskites called 2D hybrid perovskites. Compared to perovskites made of 3D crystals, these tend to be more stable, built like molecular baklava with alternating layers of metal- and carbon-based molecules. The metal-based layer, called the inorganic layer, interacts with light to produce electricity and is most efficient when its atoms align properly. The carbon-based, or organic, layer is composed of positively-charged molecules that balance the negatively-charged inorganic layer.
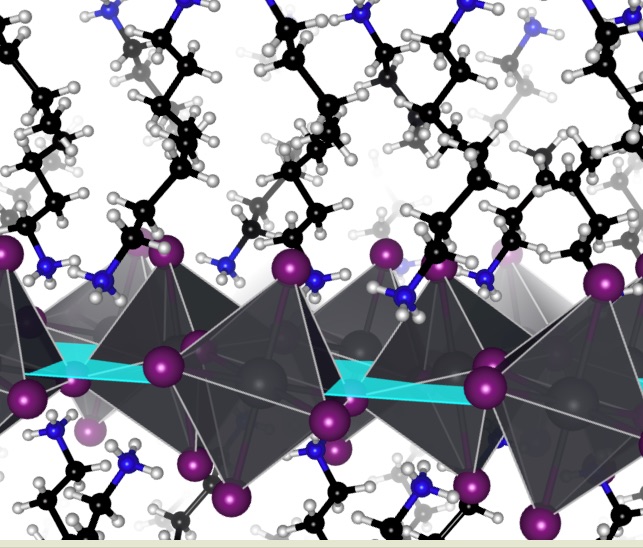
Initially, the Princeton team prepared a set of 2D perovskites with different organic molecules, studying how those molecules affected the inorganic layer’s alignment and the solar cell efficiency. In particular, they looked at a class of short, flexible organic molecules, each with a positive charge at one end. They noticed that the type of molecule influenced the structure and energy efficiency of the solar cells but didn’t exactly know why or how. They needed an atomistic insight to complement the experimental findings and hypotheses. This would help explain the system’s high performance.
So, they reached out to Rappe and Arvin Kakekhani, then a postdoc in the Rappe group, experts in using computers to model chemical interactions. “[The Princeton researchers] are very intelligent experimentalists and had great insight on the experimental level,” says Kakekhani. “But they needed knowledge and insight on the atomic, molecular level.” That’s precisely the kind of work in which the Rappe lab excels, having previously collaborated with the Loo group to model other perovskite materials in the context of rationalizing their mechanical properties.
From the current quantum mechanical calculations and charge modeling work, Kakekhani and Rappe found that the molecules in the organic layer could interact with each other, lining up in pairs or in zigzags between the metal-based layers of the perovskites.
When forming these pairs or zigzags, the organic molecules interacted less with the metal-based layer, giving the layer space to align properly and improving the performance of the resulting solar cells. The more readily the organic molecules could pair up and get out of the way of the inorganic layer, the better the efficiency of the resulting solar cell.
This observation alone offered insight into how to make better perovskites. But Kakekhani wondered whether he could find a way to capture this phenomenon in a simple value that described the interaction between the organic and inorganic layers. After testing various models, he landed on one that described how far away the interactions in the organic layer pulled the positive charge from the inorganic layer. Then he tested it to see whether it might predict how well the inorganic layer would align and how well the solar cells might perform.
Rather than fitting a model using data from the experiment, he opted to build it purely using the mathematical and physical understanding of how chemicals interact. This is known as first-principles materials modeling.
These sorts of models often struggle to accurately replicate real-world results, as they might be too simple, only considering a small subset of possible phenomena involved in a complex experiment. First-principles modeling becomes more powerful when it can give physical insight and improve the understanding of how to reduce a complex problem to a simpler one without much damage to the model’s fidelity.
In this case, Kakekhani predicted the real-life trends with surprisingly high fidelity. In mathematical terms, his model gives a coefficient of determination of >0.95, almost a perfect linear correlation. “I had never seen such a perfect correspondence between first-principles models and complex experimental observables before,” says Kakekhani. “To connect a model that sits in a computer and does not know anything about the experiment to real matter with all sorts of defects and larger scale structures—that was really surprising.”
Because this metric only needs a computer to predict solar cell performance, it could allow scientists to choose which molecules might work best in perovskites before stepping into lab, helping researchers narrow their efforts to only the most promising candidates. “There are literally millions of molecules that people could try. But it’s not so easy to make millions of solar cells,” says Rappe. “This gives people a simple scoring rule, where they could analyze whether a molecule they’re considering is likely to enhance the productivity of the solar cell.”
In the future, Rappe says these insights might also help with perovskite LEDs. If these perovskites can turn light into energy efficiently, they should be able to do something similar when turning energy to light. The groups plan to see whether the same model applies to different inorganic layers and a broader range of organic molecules, or whether other factors need consideration to accurately model the perovskite.
For now, though, the model uses one value to predict the performance of a complex solar cell, and the simplicity of the model is its strength, says Kakekhani. “Simplicity creates insight, and that insight can really create great advances in science because it goes into the nonlinear creative part of your brain. It stays there and it helps you come up with all sorts of intuitions.”
Andrew M. Rappe is the Blanchard Professor of Chemistry in the Department of Chemistry in the School of Arts & Sciences, with a secondary appointment in the Department of Materials Science and Engineering in the School of Engineering and Applied Science. He also co-directs the Vagelos Integrated Program in Energy Research at the University of Pennsylvania.
Other contributors to the work include Arvin Kakekhani, a former postdoc in the Rappe group, and Princeton University’s Yueh-Lin Loo, Xiaoming Zhao, Melissa L. Ball, and Tianran Liu.
Funding for this work came from the Department of Energy (DOE), the National Science Foundation, and the Office of Naval Research, with computational support from the Department of Defense and DOE.



