Cancer cells selectively load ‘drones’ to keep T cells from infiltrating tumors
Biologist Wei Guo and colleagues elucidate the process of sorting and loading cargo for these biological drones with implications for a more targeted and effective use of checkpoint inhibitor drugs in cancer treatment.
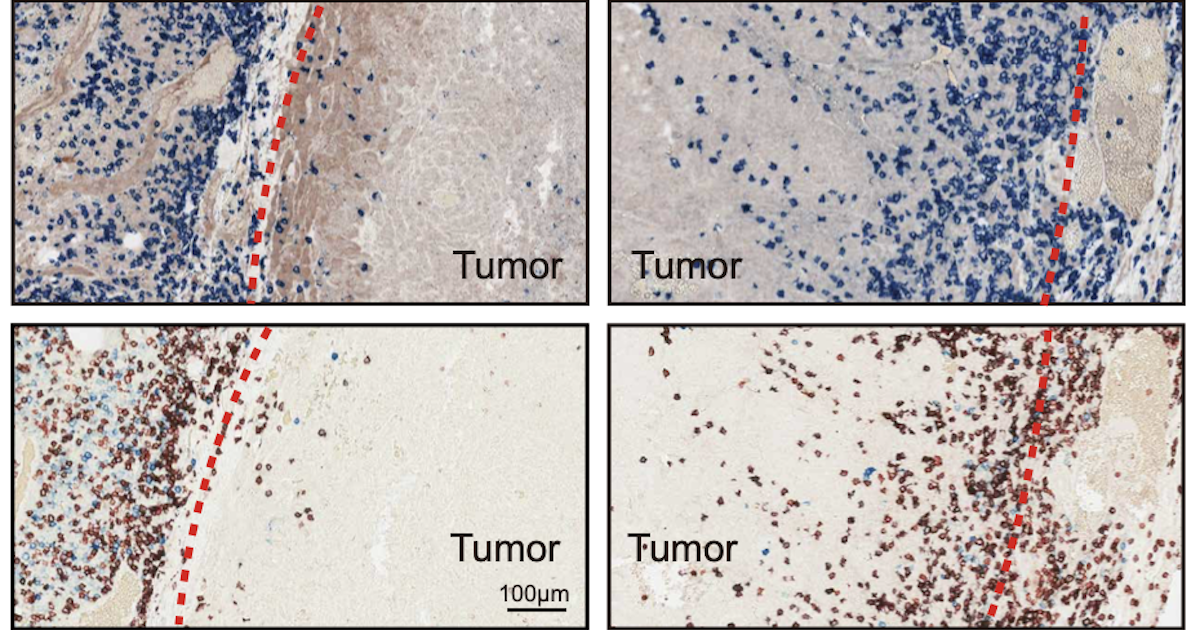
Researchers found a mechanism by which cancers keep T cells from infiltrating tumors. In areas of a tumor with low levels of a molecule called HRS with a phosphate group added, T cells could penetrate (right panels), but they were nearly blocked in areas with high levels of phosphorylated HRS (left panels). (Image: Guo lab/Nature Communications)
Checkpoint inhibitor drugs have been a gamechanger for many cancer patients. Yet around 70% don’t respond to these medications, which work by removing the “brakes” on the immune system’s response to a tumor.
Researchers have found that in some of these non-responders, the immune system’s killer T cells, which normally act to eliminate cancerous cells, cannot penetrate tumor boundaries.
In a new study, biologist Wei Guo of Penn’s School of Arts & Sciences identifies a mechanism responsible for keeping killer T cells from infiltrating a tumor. The work, which appears in the journal Nature Communications, offers a new tool to predict how a patient might respond to checkpoint inhibitor drugs.
“Infiltration of the tumor is crucial for the success of checkpoint blockade therapy,” says Guo, senior author on the work. “Because this T cell infiltration is so crucial, it’s also important to know the molecular mechanism of how it works and when it doesn’t.”
In previous research, Guo’s lab has explored how cancerous tumors secrete exosomes, small vesicles that serve as biological drones, doing battle with the immune system at sites well away from the primary tumor. That study, published in Nature in 2018, found that exosomes loaded with the protein PD-L1 caused T cells to tire before they reached the tumor, leading to an ineffective attack.
The PD-L1 protein interacts with the PD-1 protein on the surface of T cells, acting to turn off the T cells’ anti-cancer response. “If you consider exosomes as drones, then PD-L1 is the weapon loaded on the drone,” Guo says.
Many checkpoint inhibitor drugs block this interaction, which, in some cases, can keep T cells invigorated and ready for a fight. In the new work, Guo and colleagues wanted to understand how cancer cells were loading the drones with PD-L1, and whether this activity was preventing T cells from infiltrating tumors.
The researchers turned their focus to the process by which cancer cells generate exosomes, paying particular attention to a molecule called HRS, a member of a larger protein complex called ESCRT involved in sorting the “cargo” placed in exosomes. In the lab, they looked in metastatic melanoma cells. They found that frequently, ERK, an enzyme well-known to be activated in melanomas, added a phosphate group to HRS.
“We hypothesized that the tumor is regulating HRS, which has a major role in generating exosomes and uploading PD-L1 to their exosomes,” Guo says.
The researchers found that levels of phosphorylated HRS varied across tumors, negatively corresponding with the level of T cell infiltration in those areas. (Image: Guo lab/Nature Communications)
Guo and colleagues created different cancer cell lines, some of which had regular HRS, some of which had HRS with either reduced or elevated levels of phosphorylation. Mice exposed to the more highly phosphorylated HRS developed tumors that grew significantly faster than those in animals exposed to the other forms of HRS; these animals also had lower levels of T cell infiltration in their tumors.
When the researchers gave mice with highly phosphorylated HRS a checkpoint inhibitor drug that blocked PD-1, the tumors did not respond. Yet when they added an ERK inhibitor drug, the tumors shrank. Further studies showed that exosomes processed by phosphorylated HRS had cargos enriched with PD-L1, promoting resistance to the checkpoint blockade.
Indeed, when the researchers examined tissue samples of melanoma patients, different areas of the tumors had varied levels of phosphorylated HRS. Higher levels corresponded with reduced levels of T cell infiltration. “In the areas of the tumor with a lot of phosphorylation, the T cells stayed outside the border,” says Guo. “They just could not cross.”
The findings suggest that screening cancer patients’ tumors for HRS phosphorylation levels could determine whether they might respond well to checkpoint inhibitors. Further, the discoveries point to another strategy for boosting the immune system’s defenses against cancer: reducing HRS phosphorylation. ERK inhibitors are one possibility but could be toxic, says Guo. Ideally, he says, one could block phosphorylation of HRS specifically, to prevent it from loading PD-L1 to exosomes in the first place.
Guo’s group will continue investigating how tumor cells can be so selective in choosing which cargo to send out in exosomes—and how T cells, in turn, can selectively send out their own exosomes to fight back.
“It’s a clash of two titans,” Guo says.
Wei Guo is the Class of 1965 Endowed Term Professor of Biology in the University of Pennsylvania School of Arts & Sciences.
Guo’s coauthors were Penn’s Lei Guan, Bin Wu, Ting Li, Gaurav Sharma, Mingyue Li, Chin Nien Lee, Shujing Liu, Changson Yang, Lili Huang, Tatyana M. Svitkina, Lynn M. Schuchter, Giorgos C. Karakousis, Tara C. Mitchell, Youhai H. Chen, Ravi K. Amaravadi, and Xiaowei Xu; The Wistar Institute’s Lynn A. Beer, Fangping Chen, David W. Speicher, and Meenhard Herlyn; Massachusetts General Hospital’s Dennie T. Frederick, Genevieve M. Boland, and Keith T. Flaherty; China’s National Institute of Biological Sciences’ Guangcan Shao and Meng-Qui Dong; Fox Chase Cancer Center’s Kathy Q. Cai; and Oregon Health & Science University’s Gordon B. Mills. Guo was corresponding author and Guan, Wu, and T. Li were co-first authors on the study.
The study was supported by the National Institutes of Health (grants GM141832 and CA174523).
Nanoparticle blueprints reveal path to smarter medicines
New research involving Penn Engineering shows detailed variation in lipid nanoparticle size, shape, and internal structure, and finds that such factors correlate with how well they deliver therapeutic cargo to a particular destination.
A generous gift from alumni Glenn and Amanda Fuhrman brings the work of internationally acclaimed artist Jaume Plensa to the University of Pennsylvania. The latest addition to the Penn Art Collection expands Philadelphia's public art.
A massive chunk of ice, a new laser, and new information on sea-level rise
For nearly a decade, Leigh Stearns and collaborators aimed a laser scanner system at Greenland’s Helheim Glacier. Their long-running survey reveals that Helheim’s massive calving events don’t behave the way scientists once thought, reframing how ice loss contributes to sea-level rise.




{kind=link}
{kind=link}