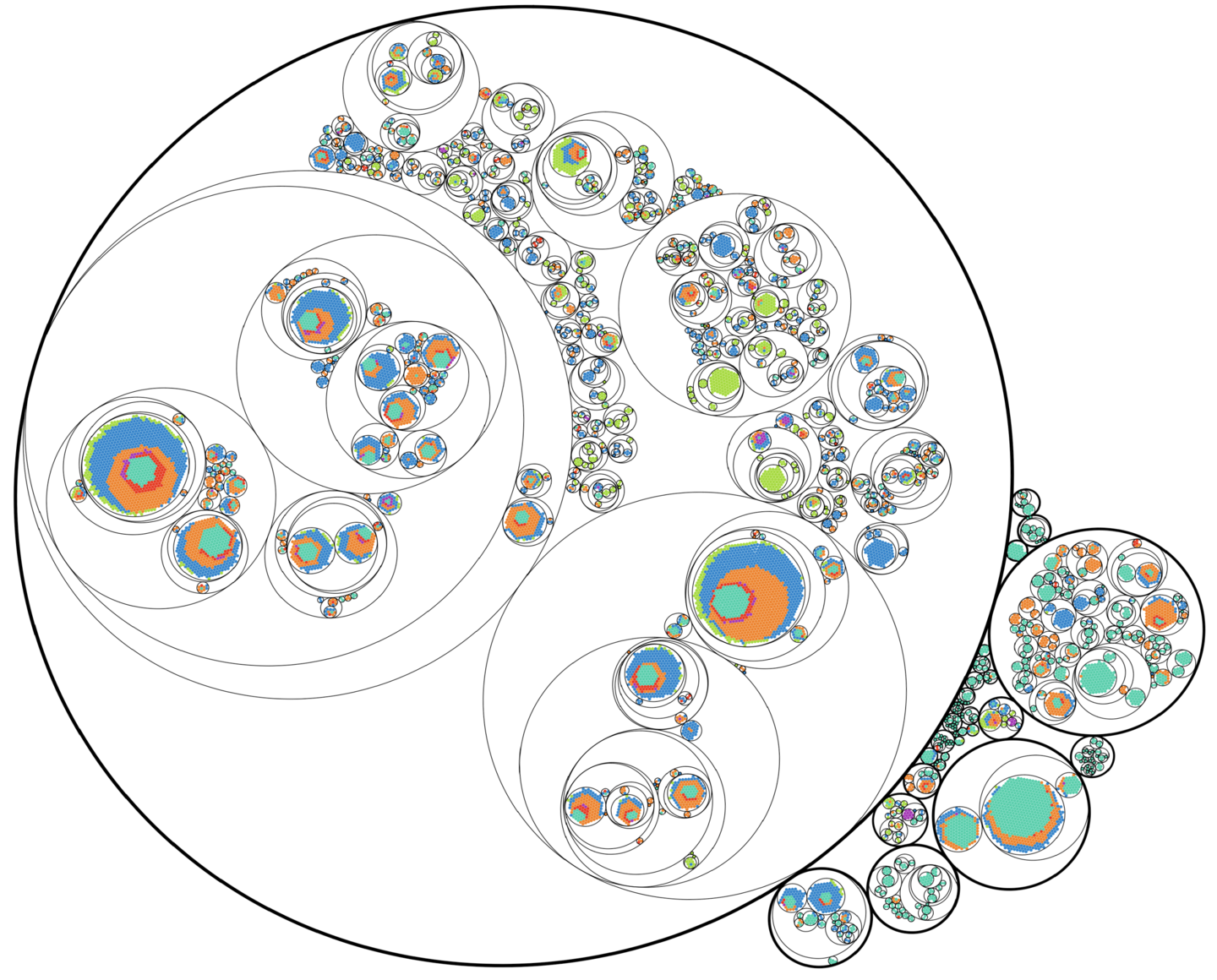
The researchers used a novel technique to precisely track the lineage of cancer cells, noting that one clone (large circle) came to dominate the metastatic cells. (Image: Courtesy of Kamen Simeonov)
In a new study, a team led by Simeonov and School of Veterinary Medicine professor Christopher Lengner has made strides toward deepening that understanding by tracking the development of metastatic cells. Their work used a mouse model of pancreatic cancer and cutting-edge techniques to trace the lineage and gene expression patterns of individual cancer cells. They found a spectrum of aggression in the cells that arose, with cells that were likely to remain in place at the primary tumor at one end and those that were more likely to move to new sites and colonize other tissues at the other end.
Of the cells that eventually became metastatic and grew in tissues and organs beyond the pancreas, the majority shared a common lineage, the researchers discovered.
“By building a precision tool for probing cancer metastasis in vivo, we’re able to observe previously inaccessible types of information,” says Simeonov. “We were able to use this lineage tracing approach to rank cells based on how metastatic they were and then relate these differences in behavior to gene expression changes.”
The group’s findings, published in the journal Cancer Cell, suggest that it’s not only genetic mutations that can drive cancer’s spread; the single-cell RNA profiling results underscore that gene expression patterns—which genes cells are turning on and off—play a key role in disease outcomes.
Fusing novel techniques
While scientists have characterized hundreds of genetic mutations associated with driving normal cells to become cancerous, they haven’t had the same success in identifying mutations that turn cancerous cells metastatic.
One possibility may be that the process is dependent on factors other than mutations, or depends on so many abnormalities grouped together that the signature is hard to resolve.
In order to better understand the biological changes that accompany metastasis, Simeonov, Lengner, and colleagues aimed to meticulously track this process, using evolving barcoding, also referred to as CRISPR lineage tracing, which enables reconstruction of cell family trees. They paired this with single-cell RNA sequencing to get a picture of the genes being turned on in each cell.
To track lineage, the researchers developed a new method employing CRISPR/Cas9 to mutagenize synthetically introduced DNA sequences, serving as cellular barcodes. These engineered cancer cells were then injected into a mouse and allowed to metastasize. While the cancer develops and spreads in the host mouse, the cellular barcodes are randomly “edited” by CRISPR/Cas9. The resulting barcode editing patterns can be used, Simeonov says, “to reconstruct phylogenetic trees of the cancer cells as they’ve proliferated and metastasized throughout the body.”
Looking at roughly 28,000 cancer cells across multiple organs of two mice, the researchers were able to see which genes each cell was turning on as the cancer spread from the pancreas to other organs and tissues. They also tracked where the cells were disseminating in the body to see if particular lineages were more likely to be metastatic than others.
“So for all of these cells, we know where they were located in the body, we have a metric for how well they metastasized, and then we also have their transcriptomes,” or catalog of RNA molecules, Simeonov says.
Spectrum of aggression
When the research team examined these pools of data together, they were surprised to find that about half of the clones, or distinct populations of cancer cells, were confined to the primary tumors.
And when they looked at the clones that had spread, they found just one dominant clone in each mouse.
“Surprisingly, despite using an aggressive cancer cell line that should be readily able to metastasize, we found that one clone dominated metastastic sites,” Simeonov says. “We were expecting more equitability between clones.”
The transcriptome profile of this dominant clone in the metastases, as well as other clones that spread from the primary tumor, were distinct from one another and from clones that stayed confined to the primary tumors. The gene expression data from this aggressive clone revealed that it had turned on genes associated with what’s known as the epithelial-mesenchymal transition (EMT), a process believed to lend cancer some of its aggressive qualities. Across the entirety of clones, the team found cells occupied different places in the EMT spectrum, from having many epithelial genes expressed to having many mesenchymal genes expressed. “Cells appeared to exist along a continuum of EMT states,” Simeonov says.
The genetic profile of the more aggressive cells turned up numerous matches with genes associated with human cancer, some of which have been predictive of reduced survival. The researchers also found that, in a particularly aggressive clone from the second mouse, a gene family associated with cancerous properties, such as cell migration and the ability to enter and exit blood vessels, was dramatically overexpressed compared to other clones.
“The expression of this gene family appeared to propagate across distinct populations and enhance the ability to metastasize in a process potentially complementary to EMT,” says Simeonov.
In future work, Simeonov, Lengner, and colleagues hope to further their studies of the process of metastasis, while also exploring new avenues for applying this lineage-tracing tool, such as examining the process of development, stem cell biology, or the regeneration of lung or intestinal tissue.
“We hope that our approach enables previously inaccessible questions to be explored and answered,” Simeonov says.
Christopher Lengner is an associate professor in the Department of Biomedical Sciences in the University of Pennsylvania School of Veterinary Medicine.
Kamen Simeonov is an M.D.-Ph.D. student in Penn’s Medical Science Training Program in the Perelman School of Medicine.
Simeonov and Lengner’s coauthors on the study were Penn’s China N. Byrns of the Perelman School of Medicine and School of Arts & Sciences and Megan L. Clark, Robert J. Nogard, and Ben Z. Stanger of the Perelman School of Medicine, as well as Beth Martin and Jay Shendure of the University of Washington and Aaron McKenna of the Dartmouth Geisel School of Medicine.
The study was supported by the National Institutes of Health (grants DK120135, HD083185, HG010152, CA023108-37, and CA168654), Blavatnik Family Fellowship in Biomedical Research, Howard Hughes Medical Institute, Allen Discovery Center for Cell Lineage Tracing, and Shipley Foundation Program for Innovation in Stem Cell Science.
Nanoparticle blueprints reveal path to smarter medicines
New research involving Penn Engineering shows detailed variation in lipid nanoparticle size, shape, and internal structure, and finds that such factors correlate with how well they deliver therapeutic cargo to a particular destination.
A generous gift from alumni Glenn and Amanda Fuhrman brings the work of internationally acclaimed artist Jaume Plensa to the University of Pennsylvania. The latest addition to the Penn Art Collection expands Philadelphia's public art.
A massive chunk of ice, a new laser, and new information on sea-level rise
For nearly a decade, Leigh Stearns and collaborators aimed a laser scanner system at Greenland’s Helheim Glacier. Their long-running survey reveals that Helheim’s massive calving events don’t behave the way scientists once thought, reframing how ice loss contributes to sea-level rise.




{kind=link}